Regulatory Affairs ist ein interdisziplinärer Bereich, der sich mit der Zulassung, Überwachung und Anpassung von Medizinprodukten und an nationale und internationale Vorschriften befasst. Dazu gehören die folgenden Punkte:
Wir helfen Ihnen, die geltenden Vorschriften und Normen für Ihr Medizinprodukt einzuhalten, sodass die Konformität Ihres Produkts stets gegeben ist.
Dabei ist ein Schwerpunkt unserer Arbeit, Medizinprodukte auf den europäischen Markt zu bringen, was bedeutet, dass wir hier unseren Kunden dabei behilflich sind, die komplexen Anforderungen der EU – Verordnung 2017/745 (kurz: MDR) zu erfüllen.
In Bezug auf Ihr Qualitätsmanagementsystem helfen wir Ihnen, die Anforderungen der harmonisierten Norm ISO 13485 an selbiges zu erfüllen. Denn mithilfe der ISO 13485 können Sie nachweisen, dass Sie in Bezug auf das QMS die Anforderungen der MDR erfüllen.
Im Bezug auf Ihr Risikomanagement helfen wir Ihnen, die Voraussetzungen der ISO 14971 “Risikomanagement medizinischer Produkte” zu erfüllen. Denn damit erfüllen Sie nicht nur die Anforderungen für das Risiomanagement in der europäischen Union, sondern zugleich auch diejenigen des nordamerikanischen Marktes.
Beim Thema Biokompatibilität helfen wir Ihnen, die Vorgaben der ISO 10993-1 zu erfüllen. Denn sowohl die MDR als auch die US-amerikanische Food and Drug Administration räumen der Biokompatibilität einen großen Stellenwert ein.
Aber auch im klinischen Produktlebenszyklus Ihres Medizinproduktes können wir Sie unterstützen. So können wir die Dokumente Clinical Development Plan, Clinical Evaluation Plan, Clinical Evaluation Report und, falls nötig, auch die Summary of Safety and Clinical Performance (SSCP) für Sie erstellen, oder aber Sie bei deren Erstellung unterstützen. Falls klinische Daten fehlen sollten, können wir Ihnen dabei helfen, klinische Prüfungen und PMCF-Studien nach der ISO 14155 durchzuführen (ggfs. zusammen mit Partnern).
Die Erstellung von Dokumenten wie Post Market Surveillance Plan (PMS-Plan), Periodic Safery Update Report (PSUR), Post-Market Clinical Follow Up (PMCF) -Plan und -Report runden unser Profil ab.
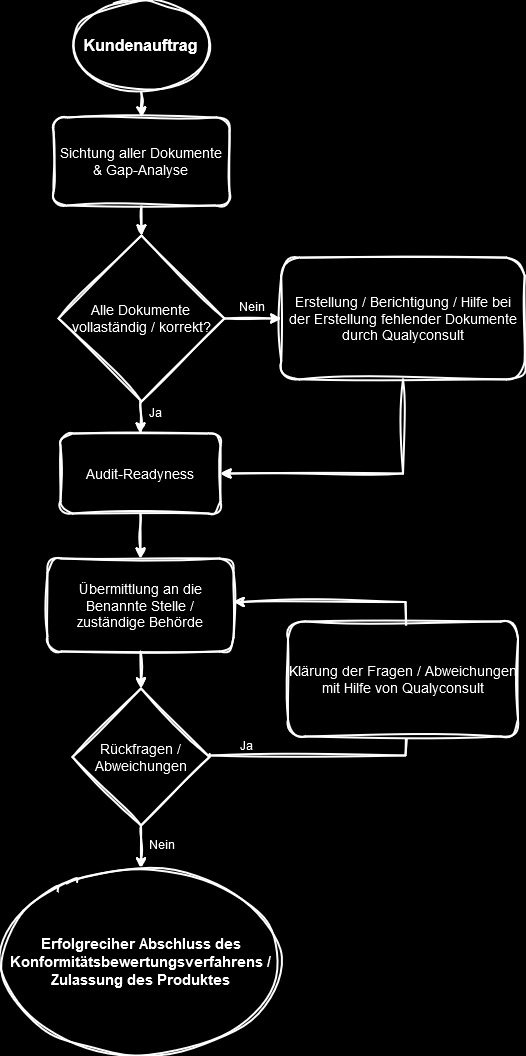
Sind alle Anforderungen der MDR und der relevanten ISO-Normen erfüllt und das Konformitätsbewertungsverfahren, in welches je nach Risikoklasse eine Benannte Stelle einzubinden ist (oder eben nicht), können Sie die Konformität Ihrer Produkte mit den Regularien erklären und Ihre Produkte auf den europäischen Markt bringen.
Denn im Gegensatz zu, z.B. den USA, werden Medizinprodukte in der EU nicht zugelassen, sondern Sie als Hersteller erklären die Konformität mit den Regularien selbst.
Falls Sie sogenannte Bestandsprodukte vermarkten, auch “legacy devices” genannt, die bereits unter der alten MDD eine Zulassung hatten, helfen wir Ihnen natürlich auch beim Übergang zur MDR und bei der effektiven Gestaltung der Übergangsfrist, die bis 2027 bzw. 2028 (je nach Risikoklasse) läuft.
Wir nutzen Cookies für ein optimales Erlebnis und zur Datenanalyse. Die Ablehnung oder der Widerruf der Einwilligung kann die Funktionalität der Website beeinträchtigen.